Genomics is a community driven data science, with existing data standards. The ability to exchange data and share results relies on a small number of common file formats; and the software tools to read, process and generate data according to these conventions. Many of the common data formats are represented in flat text files; some pre-date the internet, but they are the glue of genomics because the community supports them as de facto standards.
PetaGene Honors and Respects Community Driven Genomics Standards
PetaGene genomic data compression technology reduces the size of genomic data contained in these common file formats, without any loss of information. The compressed data is stored in a unique binary file format, but we add a transparency technology that enables the compressed data to look and interact as the original data files. Through this innovation we always present PetaGene compressed data in compliance with community accepted standards. Compressed BAM and FASTQ files appear as BAM and FASTQ files; and we provide the option to output CRAM files that are readable by any open-source tool that understands CRAM v3. PetaGene compression will never result in yet-another-file-format.
This design choice was made deliberately. By presenting our hyper-compressed data as native BAM, FASTQ, or CRAM, we honor and respect the efforts of the genomics community to maintain stable data exchange media.
Timeline of developments in genomic data formats
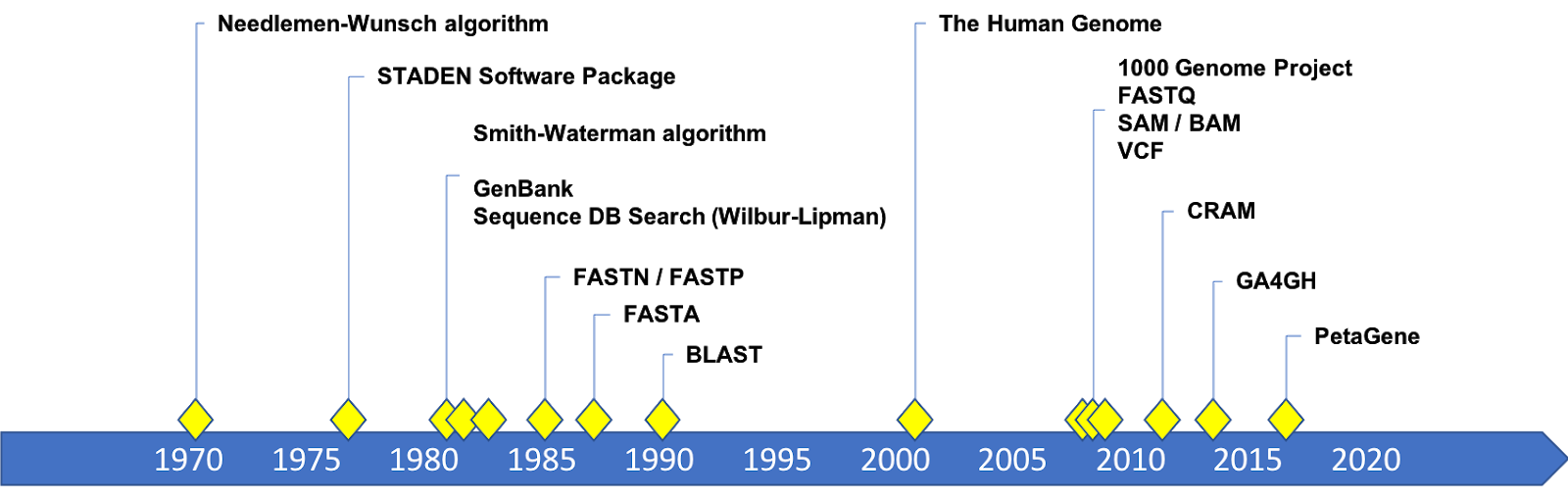
A Brief History of A C G T
The Needleman-Wunsch algorithm for global sequence alignment was published in 1970; and the Smith-Waterman algorithm for local alignment was published in 1981. Practical implementations of local alignment and search software were developed in the 1980’s. FASTP was a program for local protein sequence alignment and similarity searches published by Lipman and Pearson in 1985. Soon after, FASTA (1988) and BLAST (1990) were developed to provide fast and sensitive similarity searches for nucleotide and protein sequences. The software could read text files as input that became known as FASTA files. The structure of the text data expected by the FASTA programs became the FASTA format. This format still exists and is the format for the human genome reference sequences.
High throughput sequencing came along with the need to track the confidence of each base call in a re-sequencing result. Each sequencing technology has its own “Quality Score” and kept these scores in a separate file. Eventually sequence and quality scores were merged into one file, the FASTQ (circa 2000). There were competing versions of the format (Sanger, Solexa, and Illumina) until some convergence on quality scores occurred around 2009.
The 1000 Genomes Project drove the development of and consensus around data formats for mapped and aligned reads (SAM) and its binary, compressed form (BAM) by Heng Li and others in 2009. The 1000 Genomes Project also produced the Variant Call Format (VCF) in 2009 which captures variant information for a genome or set of genomes. CRAM is the latest addition as a format for compressed genomic data, introduced in 2011.
These formats became open specifications maintained by the 1000 Genomes Project until the Global Alliance for Genomics and Health (GA4GH) took over stewardship in 2016.

Community Standards
FASTA, FASTQ, BAM and VCF formats have persisted and become de facto standards mainly because they are simple, human readable, and an ecosystem of software tools that process these data has grown over time due to the support of many individuals and groups. There was no process to draft a standard, create an optimal representation, and approve the standard.
The informal process by which these standards have emerged may be their most important values:
- they are not the product of modern data science but they are universally understood by the community, which is more important;
- there is a legacy and diversity of tools, algorithms, and pipelines that support these formats; and
- there is a supportive group of people that will help newcomers and fix bugs when they are identified.
The adoption of new file formats or standards will happen slowly in the genomics user base due to the social and academic dynamics of the community. Due to technical and social inertia, replacing these formats is going to be very difficult for the foreseeable future. Perhaps a more formal standards process, driven by the GA4GH, will provide innovations, but this remains to be demonstrated.
At PetaGene we honor and respect these community standards. PetaGene technology provides extreme compression of genomic data without requiring the adoption of a new data structure or file format. Instead we respect the community standards by providing community compliant interfaces to the compressed data. Introducing yet another file format would provide no benefit to the community and only stifle adoption. This is why our products were engineered to present our compressed data as native FASTQ, BAM. or CRAM files.
How Does PetaGene Genomic Data Compression Support the Standards?
While we compress genomic data beyond what GZIP and CRAM can do, we present the data back as the original BAM or FASTQ files. Users and applications never see the compressed data and never need to interact with the compressed file format. Instead we employ functional interposition with the aid of an LD_PRELOAD library that provides dynamic decompression and format translation for all command line tools, applications, and pipelines. In fact, the filesystem representation of the data is also the original .bam or .fastq file names.
Our compression software also has the option to output CRAM formatted files that are created by our compressor and written to a CRAM 3.0 specification compliant file. As such, any CRAM aware tool or application can read the file without the aid of our decompression library (PetaLink). We are completely interoperable with the community standards and there is no lock-in with our compression technology.
Thus PetaGene compression technology supports the existing standards by providing users with perfectly GA4GH compliant FASTQ, BAM, or CRAM data.
What Are The Benefits of This Approach?
Supporting the community standards makes our technology immediately interoperable with all bioinformatics tools. There are no barriers to adoption and we fit right into the ecosystem of tools and technologies for processing, storing, and retrieving genomic data.
We also eliminate a major integration problem that any new format would create: being compatible with existing tools and avoiding any modification or coding to add the technology into existing workflows. By presenting data back to a tool as data it already knows, we eliminate the integration -- it just works straight out of the box. This is essentially zero-code integration.
The last reason is because BAM and FASTQ are stable formats and widely used. There is no need to propose a new format to fit our needs and expect the rest of the community to bend to our will. This does not advance bioinformatics or our business.
Open Access
Our compression technology is not open-source, it is open access. The software requires a commercial license but the basic read-back library (PetaLink) is “open access” such that it is always free and always available via petagene.com. The paid license is required for compression but not decompression. For most applications the files are compressed once to achieve storage savings and then decompressed many times. PetaLink remains free to use after the compression license has expired or depleted.
The cloud edition version of PetaLink has many additional features and requires a license.
Sustainable Commercial Support
PetaGene provides business value by making genomic data smaller and faster. Smaller data files translate into reduced storage costs and more budget for primary research activities. Faster data movement reduces processing time which accelerates discovery or provides a clinical result sooner. In our business model, we earn revenues when clients save time and money.
We charge only for compression and license fees are based on compression savings. Clients recover these fees from only a few months of storage savings. Afterwards, clients accrue 100% savings, month after month. Within one year, clients save an average of 50% in storage costs. In 5 years, these savings are over 10x of our original fee to compress the data.
We use these revenues to provide prompt and responsive support to users, fix defects, and continue to improve the product is a sustainable manner.
PetaGene also provides a fully supported, commercial implementation option for CRAM genomics data compression, should you require CRAM. Our CRAM implementation has some additional features: our reference-free compression, storage of CRAM compliant files, and transparent read-back of CRAM files to BAM with universal support of tools that don’t support CRAM. Our CRAM files can be read and processed by any tools that support CRAM v3. We will support your integration, operations, and provide technical assistance should you ever encounter problems with CRAM.
PetaSuite is a fully supported, commercial option for genomic data compression and a commercially supported implementation of CRAM. We provide clients with full warranty and support while using our software or using CRAM.
Benefits
- Extreme compression that saves money.
- Transparent read-back that eliminates integration and speeds up data transfers.
- Commercial support for PetaGene and CRAM compression workflows.
- Auditable and verifiable data integrity for lossless compression.
- Indemnity Insurance for data loss.
Conclusion
PetaGene technology is 100% compliant with community standards and GA4GH. We can provide you the best compression technology and support even if you choose to store open-source CRAM files.